日前,吉林大学物理学院马琰铭教授团队在无轨道密度泛函理论的研究中取得新进展。相关研究成果以“Nonlocal pseudopotential energy density functional for orbital-free density functional theory”为题,于2022年3月6日在线发表于《自然通讯》。
基于量子力学的第一性原理计算方法可以准确模拟出物质的微观状态以及其宏观性质。特别是Kohn-Sham密度泛函理论,由于其兼具计算精度和效率,被广泛应用于物理、化学、材料等多个学科领域, 成为最具影响力的第一性原理计算方法。然而,传统Kohn-Sham密度泛函理论的计算量随模拟体系的增大呈超线性增长,导致其仅适用于模拟原子数目小于一千个原子的系统。大多数真实材料(如合金、玻璃、复合材料等)的模拟往往需要对数万甚至百万原子系统开展研究,但传统方法的高计算成本需求,导致其难以模拟如此大尺度的系统。
无轨道密度泛函理论由于具有计算量线性标度的优势,成为开展百万原子量级大尺度材料模拟的理想候选。然而,传统观点认为:无轨道密度泛函理论仅仅依赖于系统的电子密度,只能采用近似的定域赝势方法来描述离子-电子的相互作用,而不能采用计算精度更高的非定域赝势方法,这限制了无轨道密度泛函理论的计算精度和可移植性。
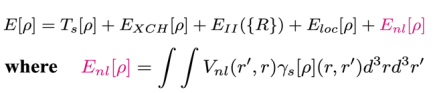
图一 非定域赝势能量密度泛函项(红色项)
吉林大学马琰铭研究团队基于密度泛函理论的基本思想,在无轨道密度泛函理论中引入了非定域赝势能量密度泛函项,据此建立了无轨道密度泛函理论新框架,使该理论能够直接采用非定域赝势计算离子-电子相互作用能。数值计算表明,基于新理论框架发展的第一性原理方法在计算精度和可移植性方面优于传统无轨道密度泛函方法。该研究为无轨道密度泛函理论的广泛应用和进一步发展打开了新的思路。
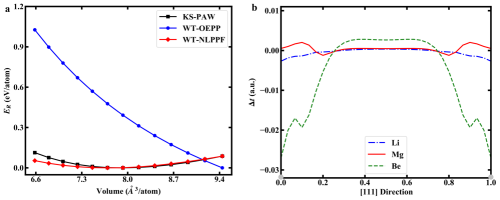
图二 以Kohn-Sham密度泛函理论为基准(KS-PAW),
无轨道密度泛函理论采用非定域赝势(NLPPF)和定域赝势(OEPP)计算Be-HCP能量-体积曲线;
Li/Mg/Be-SC的动能密度差
该研究成果的第一作者为吉林大学物理学院许强博士,吉林大学物理学院计算方法与软件国际中心和超硬材料国家重点实验室王彦超教授为本文的通讯作者。该工作得到了国家重点研发计划、国家自然科学基金、吉林大学科学技术创新研究团队和吉林大学交叉学科创新平台的资助。
谨以此文献给吉林大学物理学科七十华诞!
论文全文链接:
https://www.nature.com/articles/s41467-022-29002-3

